 The Dawn of Man. Image Credit: Movie Mezannine
The Dawn of Man. Image Credit: Movie MezannineWyoming INBRE Bioinformatics Core
Dept. of Molecular Biology
University of Wyoming
The Dawn of Man. Image Credit: Movie Mezannine
1. Put away your phone for the duration of the class
2. Do not race ahead in the tutorial.
3. Doing so compromises your learning experience.
4. You may not leave early today!
For this entire exercise, replace <inbreNNN> with your own inbre ID. Once you log onto the Teton computing environment, copy the data for today’s exercise as follows.
$ ssh <inbreNNN>@teton.uwyo.edu
$ cd /project/inbre-train/<inbreNNN>/
$ mkdir Week5_LastName && cd Week5_LastName/
$ cp -r /project/inbre-train/Week5Data/* .
$ ls -lh
This is a standard format for storing information on polymorphism across the genome. It is a n x n matrix of individuals and variants. Columns refer to individuals, whereas rows refer to variants (SNPs). Analogous to the FASTQ format, VCF also stores quality information. Files formatted to store variant calls always end in .vcf extension. For example: input.vcf. The VCF format specification is described in detail in the user manual available at http://samtools.github.io.
VCFTools is a program that can parse and manipulate the information in .vcf files. A comprehensive user manual on VCFTools usage is available here. Before we can use this program, we must load it in memory as a module.
module load gcc swset
module load vcftools
vcftools
VCFtools (0.1.14)
© Adam Auton and Anthony Marcketta 2009
Process Variant Call Format files
For a list of options, please go to:
https://vcftools.github.io/man_latest.html
Alternatively, a man page is available, type:
man vcftools
Questions, comments, and suggestions should be emailed to:
vcftools-help@lists.sourceforge.net
We have provided you with an example .vcf file for today’s exercise: human_chr15.vcf. As the name suggests, this file contains data from Homo sapiens Chromosome 15. The data was collected as part of the 1000 Genomes Project whose aim is to understand human genomic variation across the globe. This data is from the final phase of the project and contains genotypes of over 2500 individuals from 26 populations distributed globally genotyped at more than 84 million SNPs. See the map below for where those populations come from:
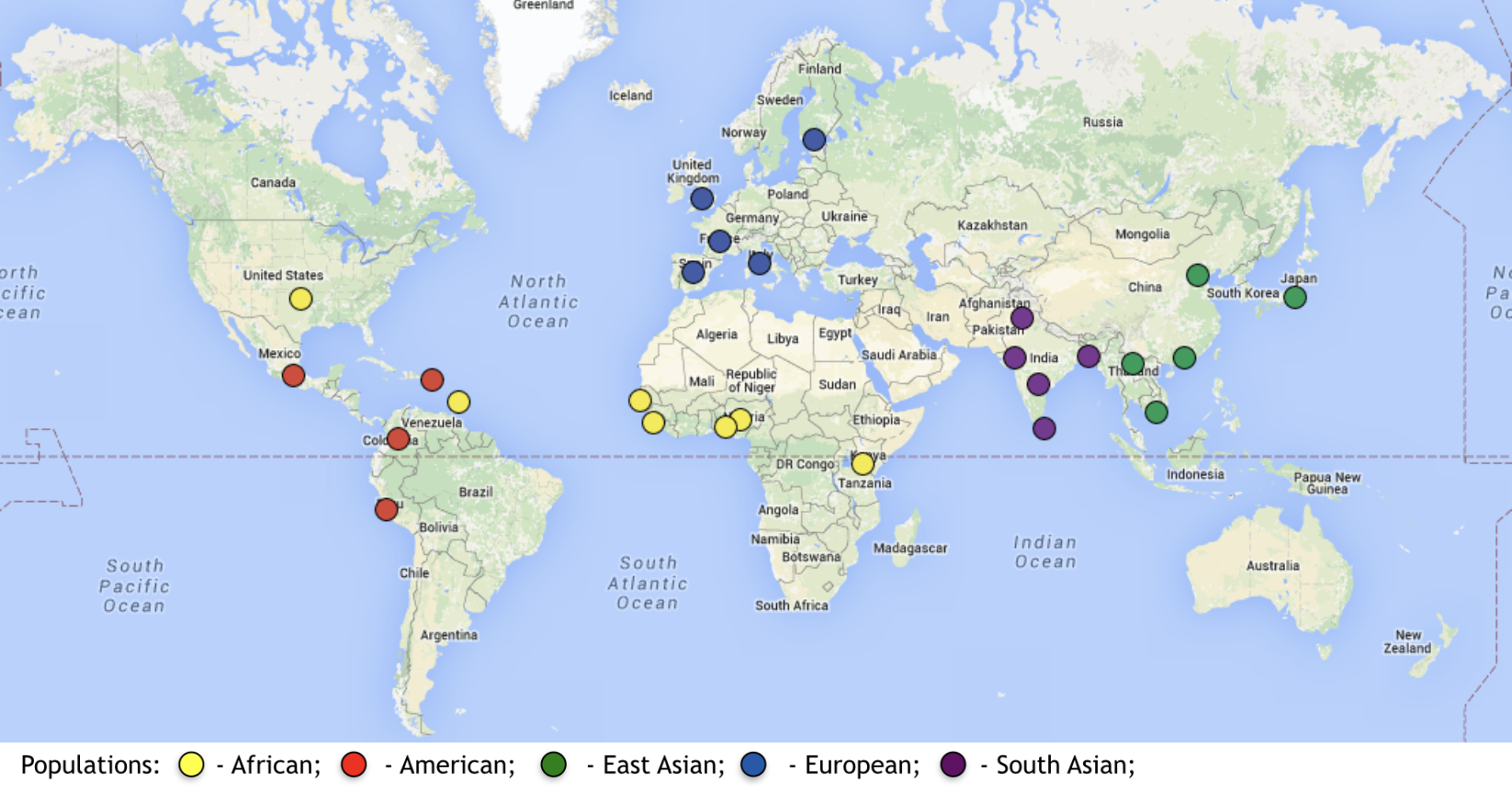 Image Credit: 1000 Genomes Project
Image Credit: 1000 Genomes Project
To make it more manageable, we have subset the original data to only include information on 16000 SNPs. You will be further subsetting this file to obtain data on one specific SNP that we are interested in. But for now, open the file in text editor and take a look at the contents. Then close the file without saving.
Note: vi that you have been using so far is synonymous with vim. Both commands work the same.
VCFTools can provide a quick summary of your data. When running without any options, VCFTools outputs summary statistics but does not make any changes to the file nor does it produce any output files.
vcftools --vcf human_chr15.vcf
VCFtools - 0.1.14
(C) Adam Auton and Anthony Marcketta 2009
Parameters as interpreted:
--vcf human_chr15.vcf
After filtering, kept 2504 out of 2504 Individuals
After filtering, kept 16003 out of a possible 16003 Sites
Run Time = 1.00 secondsrs1426654For today’s exercise, we are interested in only one SNP: rs1426654. Every documented SNP from the human genome has a rs ID associated with it. Note that the number following rs, is not the physical position of this SNP on a chromosome, but simply an identifier. You can look up the physical position of this SNP in the VCF file. For example:
Why did we pass the output of grep to cut via a |? It’s because when grep matches a pattern, it prints the entire line to the screen. Our lines here have more than 2500 columns and at this time, we are only interested in the first few columns as displayed above.
The gene SLC24A5 encodes a protein called Na/K/Ca Exchanger 5 which greatly influences skin color in humans. The leading hypothesis is that when our ancestors traveled out of Africa into Europe, the darker skin color was of no advantage in Europe’s cold climate. So eventually a new variant of the gene evolved in northern human populations which reduced the amount of skin pigmentation. By comparing the frequency of the two forms (alleles) of this gene in various human populations, we can study how populations can adapt differently when they are subject to very different environments.
rs1426654While there are more than 16,000 SNPs in our vcf file, we only need one for today’s exercise. So let’s go ahead and subset the VCF for that SNP.
vcftools --vcf human_chr15.vcf --snp rs1426654 --recode --out SLC24A5
VCFtools - 0.1.14
(C) Adam Auton and Anthony Marcketta 2009
Parameters as interpreted:
--vcf human_chr15.vcf
--out SLC24A5
--recode
--snp rs1426654
After filtering, kept 2504 out of 2504 Individuals
Outputting VCF file...
After filtering, kept 1 out of a possible 16003 Sites
Run Time = 1.00 secondsIn the above command, we extracted data for SNP rs1426654 from the file human_chr15.vcf and provided a prefix SLC24A5 for the output file. Check the output:
ls -lh
-rw-rw-r-- 1 vchhatre inbre-train 156M Nov 5 21:44 human_chr15.vcf
-rw-rw-r-- 1 vchhatre inbre-train 307 Nov 5 22:54 SLC24A5.log
-rw-rw-r-- 1 vchhatre inbre-train 30K Nov 5 22:54 SLC24A5.recode.vcfThe information that was printed to the screen was also populated inside the log file SLC24A5.log. Your main output file is SLC24A5.recode.vcf, but let’s change its name to SLC24A5.vcf for convenience.
$ mv SLC24A5.recode.vcf SLC24A5.vcf
$ ls -lh
-rw-rw-r-- 1 vchhatre inbre-train 30K Nov 5 22:54 SLC24A5.recode.vcfAs we discussed earlier, allele frequency is a measure of the relative proportion of the two (sometimes more) forms of a gene in a population. If a given SNP is not polymorphic, it will have only one allele (form). Frequency of that form will be f = 1.00 or 100%. However, if it is polymorphic, the resulting gene forms (alleles) may be present at any frquency from 0.01 to 0.99, whereas the other allele’s frequency will then be proportionately between 0.99 and 0.1. No matter their relative proportion, the frequencies of the two alleles will always add up to 1 in any population sample.
rs1426654 AllelesWe can use VCFTools to quickly estimate frequencies of the two alleles (A/G). But remember that allele frequency is a measure of population(s). During today’s exercise, we want to calculate these frequencies in each of the 26 human populations that we have data for. However, the vcf files does not contain any information on which individuals belong to which populations. Instead the population classification information is present in the folder popfiles:
$ ls -lh popfiles/
total 0
-rw-r--r-- 1 vchhatre 792 Nov 5 21:55 ESN.pop
-rw-r--r-- 1 vchhatre 816 Nov 5 21:55 STU.pop
-rw-r--r-- 1 vchhatre 768 Nov 5 21:55 PJL.pop
-rw-r--r-- 1 vchhatre 856 Nov 5 21:55 IBS.pop
-rw-r--r-- 1 vchhatre 744 Nov 5 21:55 CDX.pop
-rw-r--r-- 1 vchhatre 752 Nov 5 21:55 CLM.pop
-rw-r--r-- 1 vchhatre 512 Nov 5 21:55 MXL.pop
-rw-r--r-- 1 vchhatre 824 Nov 5 21:55 CHB.pop
-rw-r--r-- 1 vchhatre 832 Nov 5 21:55 PUR.pop
-rw-r--r-- 1 vchhatre 792 Nov 5 21:55 KHV.pop
-rw-r--r-- 1 vchhatre 824 Nov 5 21:55 GIH.pop
-rw-r--r-- 1 vchhatre 680 Nov 5 21:55 PEL.pop
-rw-r--r-- 1 vchhatre 728 Nov 5 21:55 GBR.pop
-rw-r--r-- 1 vchhatre 856 Nov 5 21:55 TSI.pop
-rw-r--r-- 1 vchhatre 792 Nov 5 21:55 LWK.pop
-rw-r--r-- 1 vchhatre 680 Nov 5 21:55 MSL.pop
-rw-r--r-- 1 vchhatre 688 Nov 5 21:55 BEB.pop
-rw-r--r-- 1 vchhatre 864 Nov 5 21:55 YRI.pop
-rw-r--r-- 1 vchhatre 840 Nov 5 21:55 CHS.pop
-rw-r--r-- 1 vchhatre 816 Nov 5 21:55 ITU.pop
-rw-r--r-- 1 vchhatre 792 Nov 5 21:55 FIN.pop
-rw-r--r-- 1 vchhatre 832 Nov 5 21:55 JPT.pop
-rw-r--r-- 1 vchhatre 488 Nov 5 21:55 ASW.pop
-rw-r--r-- 1 vchhatre 792 Nov 5 21:55 CEU.pop
-rw-r--r-- 1 vchhatre 904 Nov 5 21:55 GWD.pop
-rw-r--r-- 1 vchhatre 768 Nov 5 21:55 ACB.popEach of these popfiles contains a list of individuals that belong to that specific population. Try using the linux command cat on some of the files to see what is inside. You will notice that number of lines in each file corresponds to the number of individuals in it, with one individual listed per line.
Next, we will be writing a simple shell script to automate the estimation of allele frequencies using these population files and VCFTools. This script consumes very little computational resources, so we will not run it using sbatch. Thus you can exclude the scheduler commands that begin with #SBATCH for this script.
vim makefreq.sh
#!/bin/bash
## First load the modules
module load gcc swset vcftools
## Move to the appropriate directory
cd /project/inbre-train/<inbreNNN>/Week5_LastName/
## Make folder to store frequency output
mkdir freq
## Navigate to the folder ‘popfiles/’
cd popfiles/
## For loop
for p in *.pop
do
vcftools --vcf ../SLC24A5.vcf --keep $p --freq --out ../freq/$p
done
cd ..
Save and close the file. This script demonstrates a simple for loop which iterates over popfiles shown above and executes vcftools (to estimate allele frequencies) for only the individuals in the population file currently under iteration (Think back to our example of two populations during the lecture). Go ahead and execute it. Then look at the results.
bash makefreq.sh ## Note, it is 'bash', not 'sbatch'.
pwd
/project/inbre-train/<inbreNNN>/Week5_LastName
cd freq
ls -othr *.frq
-rw-rw-r-- 1 vchhatre 80 Oct 28 23:55 ACB.pop.frq
-rw-rw-r-- 1 vchhatre 80 Oct 28 23:55 ASW.pop.frq
-rw-rw-r-- 1 vchhatre 80 Oct 28 23:55 BEB.pop.frq
-rw-rw-r-- 1 vchhatre 66 Oct 28 23:55 CDX.pop.frq
-rw-rw-r-- 1 vchhatre 66 Oct 28 23:55 CEU.pop.frq
-rw-rw-r-- 1 vchhatre 81 Oct 28 23:55 CHB.pop.frq
-rw-rw-r-- 1 vchhatre 81 Oct 28 23:55 CHS.pop.frq
-rw-rw-r-- 1 vchhatre 80 Oct 28 23:55 CLM.pop.frq
-rw-rw-r-- 1 vchhatre 81 Oct 28 23:55 ESN.pop.frq
-rw-rw-r-- 1 vchhatre 80 Oct 28 23:55 FIN.pop.frq
-rw-rw-r-- 1 vchhatre 66 Oct 28 23:55 GBR.pop.frq
-rw-rw-r-- 1 vchhatre 81 Oct 28 23:55 GIH.pop.frq
-rw-rw-r-- 1 vchhatre 81 Oct 28 23:55 GWD.pop.frq
-rw-rw-r-- 1 vchhatre 66 Oct 28 23:55 IBS.pop.frq
-rw-rw-r-- 1 vchhatre 80 Oct 28 23:55 ITU.pop.frq
-rw-rw-r-- 1 vchhatre 82 Oct 28 23:55 JPT.pop.frq
-rw-rw-r-- 1 vchhatre 82 Oct 28 23:55 KHV.pop.frq
-rw-rw-r-- 1 vchhatre 81 Oct 28 23:55 LWK.pop.frq
-rw-rw-r-- 1 vchhatre 81 Oct 28 23:55 MSL.pop.frq
-rw-rw-r-- 1 vchhatre 80 Oct 28 23:55 MXL.pop.frq
-rw-rw-r-- 1 vchhatre 80 Oct 28 23:55 PEL.pop.frq
-rw-rw-r-- 1 vchhatre 78 Oct 28 23:55 PJL.pop.frq
-rw-rw-r-- 1 vchhatre 80 Oct 28 23:55 PUR.pop.frq
-rw-rw-r-- 1 vchhatre 80 Oct 28 23:55 STU.pop.frq
-rw-rw-r-- 1 vchhatre 81 Oct 28 23:55 TSI.pop.frq
-rw-rw-r-- 1 vchhatre 81 Oct 28 23:55 YRI.pop.frqAbove, we have truncated the output of the ls command to only include files whose names end in .frq. There are however 52 files in the freq/ folder, of which half are .frq files and the other half are .log files from VCFTools. Let’s look inside one of the population allele frequency files.
The output is self explanatory. The SNP we are studying (rs1426654) is located on Chromosome 15 at base position 48426484. Being polymorphic, it has two forms (alleles). In population LWK, there are 198 chromosomes altogether (2 homologs per individual @ 99 individuals). Finally, frequencies of both alleles are provided. Notice that the frequencies add up to 1.0 as per expectation.
In order to visualize these allele frequencies for all 26 populations, we first need to combine information from all 26 output files. Simplest way would be to use cat command.
cat *.frq > all.freq
head -n 6 all.freq
CHROM POS N_ALLELES N_CHR {ALLELE:FREQ}
15 48426484 2 192 A:0.104167 G:0.895833
CHROM POS N_ALLELES N_CHR {ALLELE:FREQ}
15 48426484 2 122 A:0.188525 G:0.811475
CHROM POS N_ALLELES N_CHR {ALLELE:FREQ}
15 48426484 2 172 A:0.534884 G:0.465116
all.freq FileFor our downstream visualization analysis, we need to get this file into correct format. You may have noticed that each allele frequency estimate has a header line. We do not need this redundant information for downstream analysis. Only one instance of the header line should be suffucient. We can selectively extract only those lines that we care about from this file. Let’s grab these lines.
sed -n '1p;0~2p' all.freq > master.freq
head master.freq
CHROM POS N_ALLELES N_CHR {ALLELE:FREQ}
15 48426484 2 192 A:0.104167 G:0.895833
15 48426484 2 122 A:0.188525 G:0.811475
15 48426484 2 172 A:0.534884 G:0.465116
15 48426484 2 186 A:0 G:1
15 48426484 2 198 A:1 G:0
15 48426484 2 206 A:0.0291262 G:0.970874
15 48426484 2 210 A:0.0190476 G:0.980952
15 48426484 2 188 A:0.718085 G:0.281915
The sed command above extracts line 1 (i.e. the first header line), and then every 2nd line from all.freq and outputs them to a new file master.freq. The output contains 27 lines (header + 1 line per population). We do not have population names in this file, but their order is alphabetical. So we should still be able to discern what frequency belongs to which population during the analysis. We need to make two more changes to master.freq. Both require opening it in vim.
This file has 6 columns, but the column header has only 5 names. Delete the string {ALLELE:FREQ} and replace it with Allele_A <tab> Allele_G. You will need to open the file in vim and then activate INSERT mode to make this change, i.e. press i
Delete the allele names A: and G: as follows. This should be done using the COMMAND mode, i.e. press ESC.
After saving and closing the file make sure it looks as expected:
$ head master.freq
CHROM POS N_ALLELES N_CHR Allele_A Allele_G
15 48426484 2 192 0.104167 0.895833
15 48426484 2 122 0.188525 0.811475
15 48426484 2 172 0.534884 0.465116
15 48426484 2 186 0 1
15 48426484 2 198 1 0
15 48426484 2 206 0.0291262 0.970874
15 48426484 2 210 0.0190476 0.980952
15 48426484 2 188 0.718085 0.281915
15 48426484 2 198 0.0252525 0.974747
popdata FileAt the base of today’s working directory (/project/inbre-train/Week5Data/), there is a file named popdata. It contains alphabetical list of population names whose lines correspond to those in allele frequency file. Check to make sure the file exists. This file also contains information on geographical locations of each of the 26 human populations we are studying. The populations are in turn grouped into 5 super populations.
pwd
/project/inbre-train/[inbreNNN]/Week5_LastName/freq
ls -lh ../popdata
-rw-r--r-- 1 vchhatre inbre-train 1.6K Oct 28 22:23 popdata
popdata with master.freqThe simplest way to accomplish this is using paste utility.
paste ../popdata master.freq > freq.df
cat freq.df
pop dist superpop lat long popname CHROM POS N_ALLELES N_CHR Allele_A Allele_G
ACB 13.19 AFR 13.1776 -59.5412 African_Carib_BBDS 15 48426484 2 192 0.104167 0.895833
ASW -8.78 AFR 36.1070 -112.1130 African_Ancestry_SW_USA 15 48426484 2 122 0.188525 0.811475
BEB 23.68 SAS 23.6850 90.3563 Bengali_in_Bangladesh 15 48426484 2 172 0.534884 0.465116
CDX 22.01 EAS 22.0088 -100.7971 Chinese_Dai 15 48426484 2 186 0 1
CEU 62.28 EUR 39.3210 -111.0937 Utah_Resid_from_NWEurope 15 48426484 2 198 1 0
CHB 23.13 EAS 39.9042 116.4074 Han_Chinese 15 48426484 2 206 0.0291262 0.970874
CHS 24.48 EAS 23.9790 113.7633 Southern_Han_Chinese 15 48426484 2 210 0.0190476 0.980952
CLM 6.24 AMR 6.2442 -75.5812 Columbian_Medellin 15 48426484 2 188 0.718085 0.281915
ESN 10.22 AFR 9.0820 8.6753 Esan_in_Nigeria 15 48426484 2 198 0.0252525 0.974747
FIN 61.92 EUR 61.9241 25.7482 Finnish_Finland 15 48426484 2 198 0.989899 0.010101
GBR 56.49 EUR 55.3781 -3.4360 Bri_England_Scotland 15 48426484 2 182 1 0
GIH 22.26 SAS 22.2587 71.1924 Guj_Ind_Houston 15 48426484 2 206 0.951456 0.0485437
GWD 13.44 AFR 13.4432 -15.3101 Gambian_WestGambia 15 48426484 2 226 0.0752212 0.924779
IBS 40.46 EUR 40.4830 -4.0876 Iberian_Spain 15 48426484 2 214 1 0
ITU 11.13 SAS 11.1271 78.6569 Telugu_Ind_UK 15 48426484 2 204 0.651961 0.348039
JPT 35.69 EAS 35.6895 139.6917 Jap_Tokyo 15 48426484 2 208 0.00480769 0.995192
KHV 14.06 EAS 14.0583 108.2772 Kinh_Vietnam 15 48426484 2 198 0.00505051 0.994949
LWK -0.02 AFR -0.0236 37.9062 Luhya_Kenya 15 48426484 2 198 0.0757576 0.924242
MSL 8.46 AFR 8.4606 -11.7799 Mende_Sierra_Leone 15 48426484 2 170 0.0882353 0.911765
MXL 23.63 AMR 23.6345 -102.5528 Mex_LA_USA 15 48426484 2 128 0.515625 0.484375
PEL -9.19 AMR -9.19 -75.0152 Peruvian 15 48426484 2 170 0.282353 0.717647
PJL 31.55 SAS 31.5546 74.3572 Punjabi_Lahore 15 48426484 2 192 0.78125 0.21875
PUR 18.22 AMR 18.2208 -66.5901 Puerto_Rican 15 48426484 2 208 0.769231 0.230769
STU 7.87 SAS 7.8731 80.7718 Sri_Lankan_Tamil 15 48426484 2 204 0.485294 0.514706
TSI 43.77 EUR 43.7711 11.2486 Toscani_Italy 15 48426484 2 214 0.995327 0.0046729
YRI 10.16 AFR 7.3775 3.9470 Yoruba_Ibadan_Nigeria 15 48426484 2 216 0.0138889 0.986111freq.df File to Your Local WorkstationRecall that in order to copy files from server to local workstation, you will first need to open a new terminal window (Cmd+Tab).